
|
HT-SAS is a web service for automatic annotation of proteins using true English words. For each protein a poll of Medline abstracts connected to homologous proteins is gathered using UniProt-Medline link. Overrepresented words are detected using binomial statistics approximation, which can be applied even if the number of abstracts is limited. HT-SAS annotations could be useful in case of poorly annotated organisms and as a complement to manually curated information sources. The service is designed for a wide range of users, including those who are not involved in protein research. The user is asked to provide protein sequence(s) in fasta format. The system automatically checks if user data is properly formatted and if it is likely to be a protein. If errors are encountered at this stage, HT-SAS allows the user to correct the data and resubmit. Otherwise a temporary file is saved and the user can adjust the mining threshold parameter. The BLAST e-value parameter is essential in this literature mining approach. It defines the level at which proteins are considered similar and therefore directly influences the number of abstracts to be analyzed. Theoretically if high values are chosen (> 1e-20) then the system recognizes even very distantly related proteins as homologous, and as such, should analyzes a larger set of abstracts. The downside is it will report information that might be somewhat "noisy". If smaller e-values are chosen, HT-SAS will find less homologs, resulting in a smaller set of analyzed abstracts. When all of the sequences are annotated a results page is displayed which shows separate tables for each protein sequence submitted. 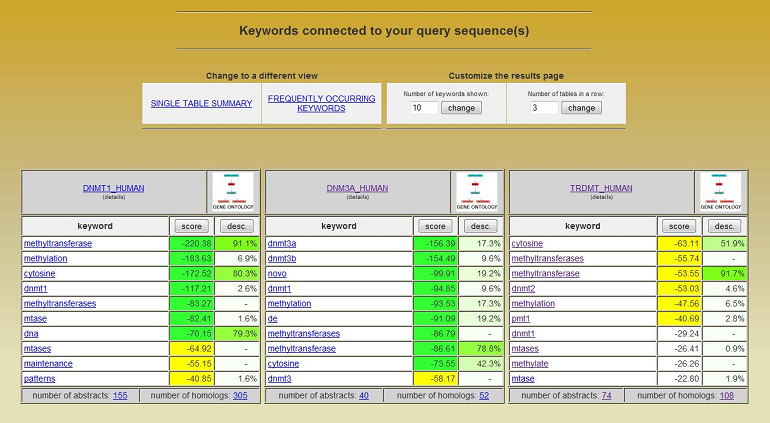 Each table has the sequence name in its title and a list of words associated with it ("annotation keywords"). Each keyword is associated with its "Score" which describes how specific and significant it is. Through these tables the user can access more specific data such as lists of (linked) publications where the word was present, lists of homologous proteins (UniProt IDs) with alignments, lists of UniProt words and Gene Ontology descriptions if present. Binomial Distribution Approximation UniProt and Medline databases are linked with each other in a many-to-many relationship (i.e. one UniProt entry can be linked with many abstracts and/or one abstract can describe numerous protein entries; a genomic paper linked with thousands of UniProt entries is such an extreme example). This is especially important considering information specificity. Therefore a normalization parameter - abstract weight - is introduced. It is calculated by dividing the number of abstract-linked proteins which are query homologues (as defined by the user with BLAST e-value parameter) and all proteins linked with this abstract. Weights are also assigned to words. They are taken directly from weighted abstracts in which they occured. The last modification to the binomial distribution was to drop the summing part. The number of analysed abstracts (N) linked to the group of homologous proteins in the majority of cases is not greater than 102. This is significantly smaller than the number of UniProt-linked abstracts which is in the order of 105. We assume that biologically significant and specific words occur in abstracts not more frequently than 10-2 - 10-4 (This is based on keywords frequency distribution, e.g. "histone" occurs in about 2500 abstracts out of more than 200 000 abstracts which are linked with UniProt). This implies a small background probability (p) which allows the following approximation shown below: 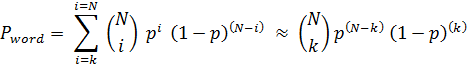
Without the need of extensive calculations (which do not significantly influence the annotation words statistics - compare: modified and not modified statistics for 100 random proteins from SGD) we are able to annotate faster and provide a more pleasant user experience with a snappier interface. More info can be found at [publication link] Please cite: [Siedlecki P., Kaczanowski S. and Zielenkiewicz P.] |

© Siedlecki P. & Kaczanowski S.